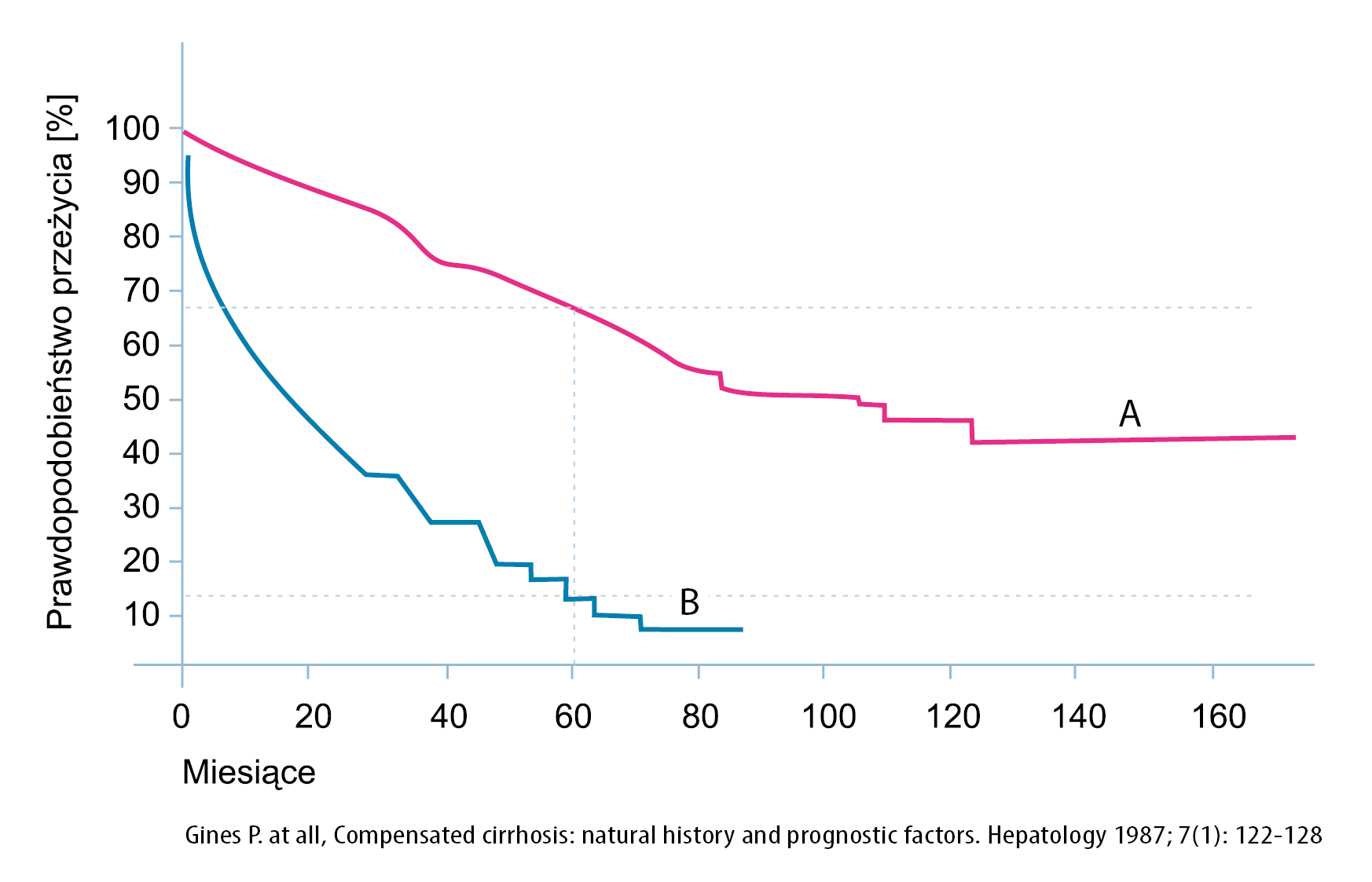
Linia A: przeżywalność od momentu rozpoznania marskości wątroby w całej populacji chorych z marskością wątroby
Linia B: przeżywalność chorych z marskością wątroby od momentu wystąpienia pierwszego objawu dekompensacji
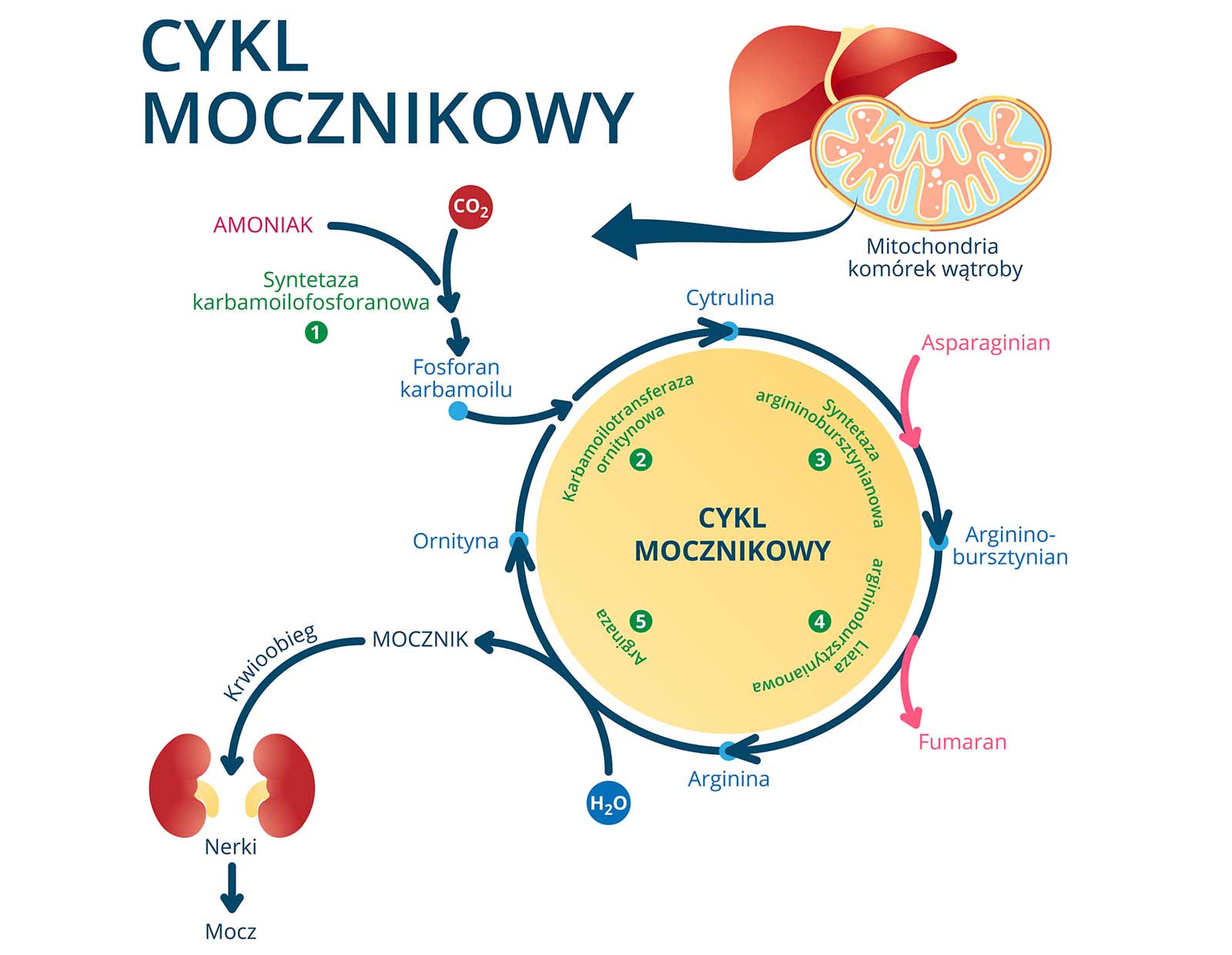
Amoniak (NH4+) + glutaminian -------------> glutamina
W warunkach wysokiego stężenia amoniaku we krwi i jego masywnego przenikania do tkanki mózgowej wspomniane mechanizmy metabolizmu amoniaku stają się relatywnie niewydolne i amoniak zaczyna toksycznie oddziaływać na OUN.
Istnieje wiele przyczyn, które w konsekwencji prowadzą do wystąpienia hiperamonemii.
Można podzielić je na trzy główne grupy. Do pierwszej grupy należą czynniki powodujące wzrost produkcji amoniaku w jelitach, do grupy drugiej – czynniki powodujące wzrost pozajelitowej produkcji amoniaku, trzecią grupę stanowią czynniki powodujące upośledzenie detoksykacji amoniaku.
najbardziej charakterystycznym objawem są problemy w radzeniu sobie z wykonywaniem wielu codziennych obowiązków w pracy i w życiu prywatnym, szczególnie jeśli dochodzi do ich nagromadzenia. Pacjenci skarżą się, że kiedyś ze wszystkim sobie radzili, a teraz te same obowiązki ich „przerastają”. Potrzebują więcej czasu i koncentracji, żeby zaplanować ich realizację i je wykonać. Szczególną trudność sprawia wykonywanie zadań wymagających koncentracji i skupienia uwagi. Mają wrażenie, że częściej zapominają. Mają też uczucie jakby spowolnienia w myśleniu i poruszaniu się. Trudności sprawia również wykonywanie niektórych działań arytmetycznych.
Kolejnym dość charakterystycznym problemem jest odwrócenie dobowego rytmu snu i czuwania, polegające na tym, że pacjent czuje się senny w ciągu dnia (co dodatkowo utrudnia wywiązywanie się z obowiązków), natomiast ma problemy ze snem w nocy.
Pacjenci zauważają zmiany nastroju – czasami są niewspółmiernie do sytuacji pobudzeni i radośni, ale częściej zdarza się, że są przygnębieni, drażliwi, odczuwają lęk lub też mają wahania nastroju.
Niekiedy obserwuje się dyskretne zaburzenia neurologiczne, takie jak pojedyncze drżenia mięśni, mniej lub bardziej niewyraźne pismo oraz zaburzenia koordynacji wzrokowo-ruchowej (ataksja), które można zaobserwować podczas wykonywania próby palec-nos. Zaburzenia te nasilają się, przy wykonywaniu tej próby z zamkniętymi oczami.
Jednakże największym problemem w początkowych stadiach choroby (stopień 0. i stopień 1.) jest tzw. wydłużenie czasu reakcji na bodźce, co ma zasadnicze znaczenie w przypadku prowadzenia pojazdów lub obsługiwania maszyn i urządzeń w ruchu.
Ponadto u chorych występuje upośledzona zdolność:
U chorych z encefalopatią wątrobową obserwuje się wydłużenie czasu reakcji, podobnie jak po spożyciu alkoholu *
* Kircheis G., Knoche A., Hilger N. et al. Hepatic encephalopathy and fitness to drive. Gastroenterology 2009; 137: 1706-1715
objawy psychiczne nasilają się. Chorzy zaczynają mieć zauważalne zaburzenia świadomości określane jako przymglenie. Mogą wystąpić nieznaczne zaburzenia orientacji odnośnie do czasu i miejsca. U chorych obserwuje się znaczne spowolnienie psychiczne i ruchowe, zmiany osobowości, nieadekwatne zachowania. Chorzy odczuwają lęk i niepokój. Nasilają się zaburzenia pamięci, wykonywanie nawet prostych działań arytmetycznych staje się bardzo utrudnione. Drżenia mięśniowe, dotychczas pojedyncze, wyraźnie się nasilają. Występuje drżenie grubofaliste o charakterze trzepotania (flapping tremor, asterixis) - nierytmiczne, asymetryczne opadanie w trakcie zależnej od woli próby utrzymania kończyny w jednej pozycji. Najlepiej można je zaobserwować, prosząc pacjenta o wyciągnięcie przed siebie kończyn górnych i grzbietowe zgięcie dłoni.
Nasila się ataksja. Pojawiają się trudności w wypowiadaniu słów (dyzartria), a pismo staje się nieczytelne. W tym stadium pacjent musi już być intensywnie leczony w warunkach szpitalnych.
pogłębiają się zaburzenia świadomości – występują znaczne zaburzenia orientacji w czasie i przestrzeni, senność, splątanie, jednakże pacjent reaguje jeszcze, w ograniczonym stopniu, na bodźce głosowe. Pojawiają się wyraźne zaburzenia psychiczne – pacjent może odczuwać silny lęk, reagować niepohamowanym gniewem lub mieć urojenia. Zaburzeniom psychicznym towarzyszą poważne objawy neurologiczne – odruchy patologiczne, wygórowane odruchy ścięgniste, skurcze kloniczne, oczopląs, objawy pozapiramidowe.
określany jest mianem śpiączki wątrobowej. Pacjent jest nieprzytomny, nie reaguje na żadne bodźce, w tym także na bodźce bólowe. Źrenice są szerokie, nie reagują na światło. Może wystąpić sztywność odmóżdżeniowa. W większości przypadków po pewnym czasie następuje śmierć.
Ze względu na opisane powyżej zaburzenia, które w znacznym stopniu pogarszają jakość życia chorych w początkowych stadiach encefalopatii lub wręcz stanowią zagrożenie życia w stadiach bardziej zaawansowanych, bardzo ważne jest jak najszybsze rozpoczęcie odpowiedniego leczenia.
Podstawą rozpoznania encefalopatii wątrobowej
w praktyce lekarza pierwszego kontaktu jest
wywiad i badanie podmiotowe.
W niektórych przypadkach może zachodzić konieczność przeprowadzenia bardziej zaawansowanej diagnostyki.
W tym celu pomocne może okazać się wykonanie następujących badań dodatkowych:
Podstawowy cel leczenia:
zmniejszenie stężenia amoniaku we krwi.
W procesie terapeutycznym należy zawsze uwzględnić:
Podjęcie działań mających na celu zwiększenie metabolizmu amoniaku jest korzystne i wskazane w każdej sytuacji, bez względu na przyczynę, która doprowadziła do hiperamonemii.
Podstawowym lekiem stosowanym w tym celu jest asparaginian ornityny (L-ornithine L-aspartate, LOLA).
Mechanizm działania asparaginianu ornityny:
Stymuluje cykl mocznikowy w hepatocytach
Aktywuje syntetazę glutaminową
Dostępne produkty lecznicze:
Dawkowanie: Należy podkreślić, że skuteczne są tylko odpowiednio wysokie dawki LOLA (!)
Dokonując wyboru produktu leczniczego, należy zawsze zwrócić uwagę na zawartość substancji czynnej, czyli asparaginianu ornityny, ponieważ na rynku dostępne są także produkty (suplementy diety, nie leki!) zawierające bardzo małe ilości LOLA (np. produkt Hepatil® w postaci tabletek zawierających zaledwie 100 mg asparaginianu ornityny). Należy zauważyć, że aby zastąpić np. 1 saszetkę leku Hepa-Merz®3000 (3g LOLA) suplementem diety Hepatil® (100 mg), należałoby podać 30 tabletek produktu Hepatil (!)
Szczegółowe informacje dotyczące wskazań, przeciwwskazań, działań niepożądanych dotyczących leku Hepa-Merz® oraz Hepa-Merz® 3000 znajdą Państwo w Charakterystyce Produktu Leczniczego.
Ponieważ amoniak w większości powstaje w jelitach jako produkt metabolizmu białka, a do jego nadmiernego wytwarzania dochodzi w przypadku zaburzeń składu bakteryjnej flory jelitowej, wydaje się zasadne stosowanie leków normalizujących skład flory jelitowej.
W praktyce w tym celu wykorzystuje się antybiotyki niewchłaniające się z przewodu pokarmowego.
Obecnie najczęściej stosowana jest rifaksymina (np. produkt Xifaxan® lub Tixteller®). Jest to niewchłaniający się z przewodu pokarmowego antybiotyk o szerokim spektrum działania bakteriobójczego (aktywny wobec bakterii G+, G-, tlenowych i beztlenowych). Poprzez redukcję patologicznej flory bakteryjnej przewodu pokarmowego zmniejsza wytwarzanie amoniaku.
Rifaksymina powinna być podawana w dawce 3 x 400mg (Xifaxan®) lub 2 x 550mg (Tixteller®).
W encefalopatii wątrobowej zaleca się długotrwałe podawanie antybiotyku, nawet przez 6 miesięcy codziennie.
Oczywiście stosowanie rifaksyminy, jak również innych niewchłaniajacych się z przewodu pokarmowego antybiotyków, jest uzasadnione jedynie w przypadku występowania zaburzeń bakteryjnej flory jelitowej. Nie ma natomiast znaczenia i uzasadnienia, jeśli występują inne przyczyny wzrostu wytwarzania amoniaku lub amoniak jest wytwarzany w prawidłowych ilościach, a przyczyną hiperamonemii jest zaburzony jego metabolizm (np. na skutek niewydolności wątroby) – patrz rozdział „PATOGENEZA – Przyczyny prowadzące do hiperamonemii”).
Szczegółowe informacje dotyczące wskazań, przeciwwskazań, działań niepożądanych dotyczących leku Xifaxan® oraz Tixteller® znajdą Państwo w Charakterystyce Produktu Leczniczego.
W celu zmniejszenia wytwarzania amoniaku w jelitach, jak również w celu skrócenia czasu jego wchłaniania (skrócenie czasu zalegania mas kałowych), stosowana jest laktuloza – dwucukier niewchłaniający się z przewodu pokarmowego.
Laktuloza rozkładana jest przez bakterie jelitowe do kwasu mlekowego i octowego, co powoduje:
- obniżenie pH – co z kolei prowadzi do zwiększonego namnażania bakterii acidofilnych i zahamowania namnażania bakterii proteolitycznych, czego efektem jest zmniejszone wytwarzanie amoniaku. Obniżone pH zwiększa ponadto wydalanie amoniaku z kałem
- działanie osmotyczne i stymulujące perystaltykę – co poprzez przeciwdziałanie przeczyszczające zwiększa usuwanie amoniaku z jelit.
Laktuloza powinna być stosowana początkowo w dawce 3-4 x 30-45ml, a następnie w dawkach dobieranych indywidualnie, tak, aby uzyskać 3 luźne stole na dobę.
Decydując się na długotrwałe stosowanie laktulozy, należy jednak pamiętać o jej działaniach niepożądanych oraz przeciwwskazaniach do stosowania:
Przeciwwskazania:
Działania niepożądane: